We have significant interest in using state-of-the-art proteomics approaches to answer diverse questions in cancer research.
In the early years of the lab, using a combination of "omics" approaches, including RNA-seq, translational profiling via ribosome profiling, quantitative proteomics, and functional genomics, we initially found that very few proteins are upregulated during rapid cell death induced by the proteasome inhibitor bortezomib, a first-line therapeutic administered to almost all myeloma patients. We developed a quantitative model to relate absolute protein molecules per cell to mRNA transcript abundance in the context of drug treatment (Liu and Huang et al., Cell Systems (2017)).
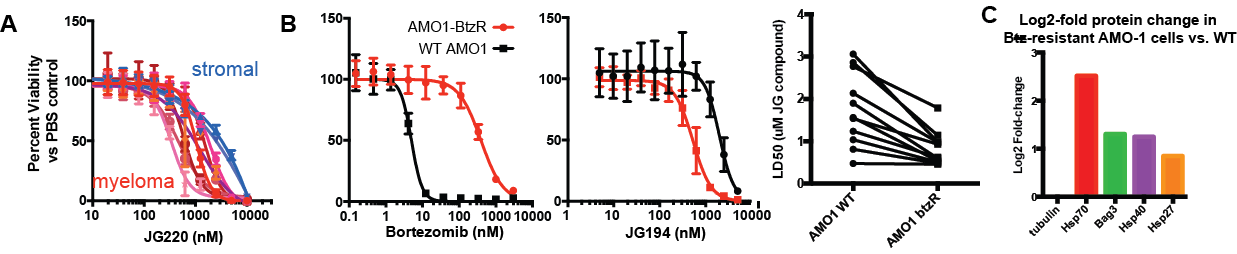
We have further used pulsed-SILAC proteomics to reveal an interaction between proteasome inhibitor resistance and vulnerability to mitochondrial perturbation, in collaboration with Jason Gestwicki at UCSF (Ferguson et al., Cell Chem Biol (2022)). These results validate HSP70 inhibition as a promising strategy to overcome PI resistance.
In collaboration with Jay Debnath's lab at UCSF, we have used TMT, data independent acquisition, and proximity labeling proteomics to evaluate novel mechanisms of secretion via the autophagy pathway (Leidal et al, Nat Cell Biol (2022)) and the role of autophagy in breast cancer metastasis (Mondal et al, Nat Cell Biol (2025)).
In collaboration with the group of the late Felix Feng, we used DIA proteomics to evaluate the role of ATG9A in antibody and CAR macrophage-directed anti-tumor therapy (Liu et al, Nat Commun (2025)) and interactomics approaches to evaluate the role of PTGES3 in androgen receptor-mediated prostate cancer therapy (Li et al, BioRxiv (2025)).
We also have several other ongoing collaborations regarding cell surface proteomics in a variety of biological systems.